27.01.2023


Główne tezy
- Zgodnie z art. 68 Konstytucji Rzeczypospolitej Polskiej każdemu obywatelowi przysługuje prawo do ochrony zdrowia, a obowiązkiem państwa jest zapewnienie równego dostępu do świadczeń opieki zdrowotnej finansowanej ze środków publicznych.
- Wypracowany na przestrzeni lat system refundacji leków stanowi, obok innych rozwiązań, odpowiedź na konstytucyjne zobligowanie państwa do równego dostępu do świadczeń opieki zdrowotnej finansowanej ze środków publicznych.
- Polityka Lekowa Państwa na lata 2018-2022 wskazywała, że wyzwaniem, ograniczającym dostęp do leków, nadal jest czas pomiędzy pojawieniem się leku, a jego praktyczną dostępnością.
- W przypadku leków refundowanych praktyczna dostępność leków oznacza dostępność na rynku leków objętych refundacją. Obecnie obowiązujące regulacje prawne znacznie opóźniają tę dostępność.
- W aktualnym stanie prawnym proces refundacyjny obejmuje złożenie wniosku o refundację do Ministerstwa Zdrowia. Następnie Agencja Oceny Technologii Medycznych i Taryfikacji dokonuje oceny złożonego wniosku, a Prezes AOTMT, na podstawie stanowiska Rady Przejrzystości), wydaje stosowną rekomendację. Po negocjacjach z Komisją Ekonomiczną wydawana jest decyzja refundacyjna.
- Już w Polityce Lekowej Państwa na lata 2018-2022 wskazywano na konieczność zwiększenia przejrzystości podejmowanych decyzji refundacyjnych oraz poziomu zaufania w dialogu pomiędzy uczestnikami systemu.
- Aktualnie toczą się prace nad projektem nowelizacji ustawy refundacyjnej, który stanowi pierwszą tak poważną próbę zmiany ustawy refundacyjnej.
- Upływ 11 lat obowiązywania ustawy refundacyjnej pokazał niewątpliwie braki i niedociągnięcia, których ustawodawca nie przewidział w toku prac nad ustawą oraz wady przyjętych rozwiązań.
- Kluczowe z punktu widzenia racjonalizacji postepowania refundacyjnego są proponowane zmiany art. 25 i art. 11 ustawy refundacyjnej. Należy je rozpatrywać w kontekście tak zwanych „powiązań patentowych”, czyli uzależnienia decyzji o rejestracji lub refundacji leku równoważnego (generyka lub biologicznego równoważnego) od statusu patentowego leku referencyjnego.
- W przypadku wejścia w życie art. 11 ust. 1a w proponowanym brzmieniu złożenie wniosku o objęcie refundacją leku generycznego w trakcie trwania ochrony patentowej lub wyłączności rynkowej obejmującej wnioskowany produkt powodowałoby wydanie przez Ministra do spraw zdrowia decyzji o odmowie wszczęcia postępowania.
- Postępowanie refundacyjne od chwili złożenia wniosku o objęcie refundacją do momentu wydania decyzji o refundacji zajmuje przeciętnie od 6 do 12 miesięcy, a w myśl art. 11 ust. 1a możliwość skutecznego złożenia wniosku o objęcie refundacją generycznego produktu leczniczego pojawi się dopiero po wygaśnięciu ostatniego z reżimów ochronnych leku oryginalnego.
- W efekcie tej możliwość refundacji tańszego zamiennika leku oryginalnego zostanie znacznie przesunięta w czasie. Dlatego też należy wycofać się z proponowanej w tym brzmieniu regulacji.
- Racjonalność wykorzystywania środków publicznych zakłada, iż refundowane powinny być te produkty, które mają udowodnioną skuteczność i bezpieczeństwo stosowania, a relacja efekt terapeutyczny – koszt terapii jest korzystniejszy w porównaniu z dostępnymi wariantami terapii.
- Funkcjonowanie powiązań patentowych w systemie refundacyjnym skutkuje licznymi negatywnymi konsekwencjami, w tym na polu konkurencji firm farmaceutycznych, budzi też wątpliwości natury prawnej. Kluczowe pozostaje jednak opóźnianie faktycznej dostępności leku, który jest odpowiednikiem leku dotychczas dostępnego i refundowanego, jednak jest tańszy. Stanowi to nieracjonalne wydatkowanie środków budżetowych.
- Poprzeć należy proponowane już w doktrynie rozwiązanie w postaci mechanizmu umożliwiający występowanie z wnioskiem o decyzję refundacyjną w ciągu określonego czasu przed upływem ostatniego z reżimów ochronnych, obejmujących dany produkt leczniczy.
- Postępowanie refundacyjne – jak jest?
Zgodnie z art. 68 Konstytucji Rzeczypospolitej Polskiej[1] każdemu obywatelowi przysługuje prawo do ochrony zdrowia, a obowiązkiem państwa jest zapewnienie równego dostępu do świadczeń opieki zdrowotnej finansowanej ze środków publicznych. Regulacja ta leży u podstaw szeregu działań legislacyjnych i politycznych podejmowanych przez państwo w celu zabezpieczenia i ochrony zdrowia jego ludności. Wypracowany na przestrzeni lat system refundacji leków – a więc możliwość partycypowania w pokrywaniu kosztów terapii częściowo przez chorego a częściowo przez państwo stanowi, obok innych rozwiązań, w tym przede wszystkim dostępu do publicznej służby zdrowia, odpowiedź na konstytucyjne zobligowanie państwa do równego dostępu do świadczeń opieki zdrowotnej finansowanej ze środków publicznych.

Fakt, że państwo w pewnym stopniu finansuje opiekę zdrowotną, podejmuje działania na rzecz jej usprawniania, upowszechniania i dofinansowania nie oznacza jednak, że wszystkie funkcjonujące w ramach tych systemów rozwiązania są właściwe, racjonalne i efektywne. Nie ulega wątpliwości, że polska polityka zdrowotna, w tym w zakresie polityki lekowej a co za tym idzie refundacyjnej – wymaga usprawnienia.
I.I. Polityka Lekowa Państwa 2018-2022
Ponieważ nowa polityka lekowa państwa na rok 2023 i kolejne nie została jeszcze opracowana i przyjęta, za obowiązującą na dzień dzisiejszy należy przyjąć politykę realizowaną dotychczas. Zgodnie z Polityką Lekową Państwa 2018-2022 (dalej jako: PLP), „celem strategicznym polityki lekowej w odniesieniu do dostępności refundacyjnej jest systematyczna poprawa stanu zdrowia populacji, dzięki optymalizacji wydatków publicznych zapewniających możliwie najszerszy dostęp do skutecznych, bezpiecznych i kosztowo-efektywnych terapii”[2].
W kontekście dostępności refundacyjnej w PLP na lata 2018-2022 zdefiniowano do osiągnięcia kilka celów, w tym m.in. poprawę efektywności wykorzystania środków publicznych w celu osiągnięcia jak najlepszego efektu zdrowotnego, czy systematyczne poszerzanie katalogu terapii o udowodnionej skuteczności w ramach realizowanego budżetu. Odnośnie procesu refundacyjnego wskazano cele w postaci zwiększenia przejrzystości podejmowanych decyzji refundacyjnych oraz poziomu zaufania w dialogu pomiędzy uczestnikami systemu.
Systematyczne poszerzanie katalogu terapii o udowodnionej skuteczności w ramach realizowanego budżetu nastąpić miało poprzez podejmowanie działań w celu skrócenia czasu na refundację skutecznych leków, w szczególności poprzez minimalizowanie obciążeń administracyjnych, wczesny dialog z firmami farmaceutycznymi, prowadzenie horizon scanning (tj. opracowanie i wdrożenie systemu monitorowania leków będących w trakcie badań klinicznych) i zaproponowanie rozwiązań prawnych umożliwiających wczesny dostęp do innowacyjnych i skutecznych terapii.
Wśród powyższych w szczególności warty uwagi jest mechanizm horizon scannning, który pozwala na podejmowanie decyzji w oparciu o szerszy zakres informacji – uwzględniając potencjalne nowe technologie, które w niedługim czasie pojawią się na rynku, a dla których pożądane będzie zapewnienie finansowania. Uzupełnieniem tego procesu powinno być monitorowanie leków, które w najbliższym czasie będą traciły ochronę patentową.
Natomiast odnośnie samego procesu refundacyjnego, Polityka Lekowa Państwa na lata 2018-2022 postulowała zwiększenie przejrzystości podejmowanych decyzji refundacyjnych oraz poziomu zaufania w dialogu pomiędzy interesariuszami. Miało to nastąpić poprzez zapewnienie realnego dialogu pomiędzy decydentami, a uczestnikami systemu (przy uwzględnieniu szczególnej roli pacjentów, nie tylko w zakresie finansowania poszczególnych leków, ale i poprawy funkcjonowania systemu refundacyjnego), równoważenie przejrzystości procesów decyzyjnych z zapewnieniem poufności informacji wrażliwych, takich jak ceny efektywne leków oraz szczegółowe warunki instrumentów dzielenia ryzyka, realizowanie zadań w stałych i czytelnych ramach organizacyjnych w celu budowania zaufania uczestników systemu do systemu ochrony zdrowia i organów administracji publicznej, a także wdrażanie zmian przepisów niebudzących wątpliwości co do swojego celu i interpretacji oraz wypracowywanie nowych rozwiązań poprzez dialog społeczny (w tym debata nad uwagami ekspertów).
Niezależnie od powyższego, w PLP wskazano, że wyzwaniem, ograniczającym dostęp do leków, nadal jest czas pomiędzy pojawieniem się leku, a jego praktyczną dostępnością. Może to być związane z opóźnieniem rejestracji w Polsce przez podmiot odpowiedzialny lub opóźnionym wprowadzeniem na polski rynek leku już zarejestrowanego, wydłużonym okresem od wprowadzenia leku na rynek do złożenia wniosku o objęcie refundacją, czasem trwania procedury o objęcie refundacją oraz jej późniejszego wdrożenia[3].
W przypadku leków refundowanych praktyczna dostępność leków oznacza dostępność na rynku leków objętych refundacją. Obecnie obowiązujące regulacje prawne znacznie opóźniają tę dostępność – jest jednak szereg rozwiązań, których wprowadzenie mogłoby to zmienić.
I.II. Proces refundacji leku w aktualnym stanie prawnym
Leki refundowane to grupa dopuszczonych do obrotu preparatów leczniczych, które mogą być sprzedawane za obniżoną ceną lub wydawane pacjentom nieodpłatnie.
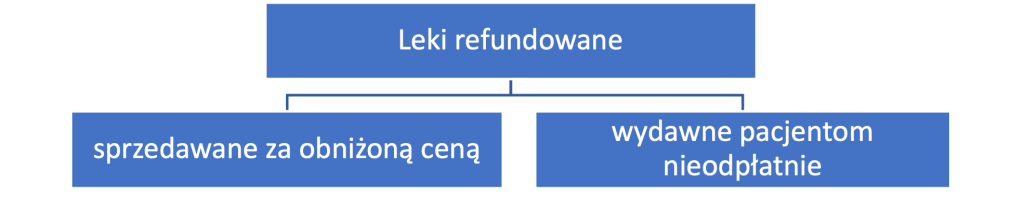
Są one objęte urzędową ceną zbytu, czyli ceną zbytu leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego ustaloną w decyzji administracyjnej o objęciu refundacją, uwzględniającą należny podatek od towarów i usług[4]. Leki te znajdują się w wykazie publikowanym przez Ministerstwo Zdrowia, który aktualizowany jest co 2 miesiące[5]. Opracowując listy leków oraz innych produktów (oprócz leków refundacji podlegają niektóre wyroby medyczne oraz środki spożywcze specjalnego przeznaczenia żywieniowego) objętych refundacją Minister Zdrowia winien brać pod uwagę skuteczność terapeutyczną, koszt oraz bezpieczeństwo każdego objętego lub nie objętego refundacją produktu leczniczego.
W aktualnym stanie prawnym proces refundacyjny, czyli proces objęcia konkretnego leku (lub innego środka) refundacją można podzielić na kilka etapów. Całość reguluje tzw. ustawa refundacyjna, czyli ustawa z dnia 12 maja 2011 r. o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych (Dz. U. z 2022 r., poz. 2555 tj.j. z późn. zm.). W pierwszej kolejności firma farmaceutyczna chcąca, aby jej produkt został objęty refundacją powinna złożyć wniosek o refundację do Ministerstwa Zdrowia. Następnie Agencja Oceny Technologii Medycznych i Taryfikacji (dalej jako: AOTMT) dokonuje oceny złożonego wniosku, a Prezes AOTMT, na podstawie stanowiska Rady Przejrzystości), wydaje stosowną rekomendację.
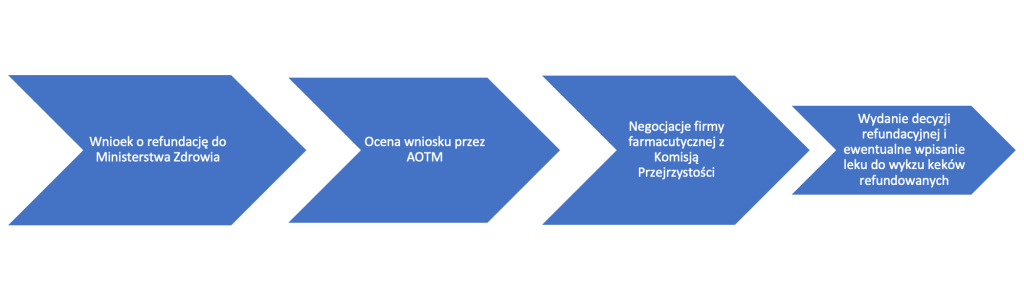
W przypadku pomyślnego zakończenia dotychczasowej procedury następują negocjacje cenowe, pomiędzy firmą farmaceutyczną a Ministerstwem Zdrowia (negocjacje powierzane są Komisji Ekonomicznej). Po wydaniu decyzji refundacyjnej przez Ministerstwo Zdrowia następuje wpisanie leku do wykazu leków refundowanych w ramach programów lekowych.
I.III. Wniosek refundacyjny
Proces refundacyjny rozpoczyna złożenie wniosku refundacyjnego. Wniosek ten stanowi pierwszą i podstawową dokumentację, jaką składa wnioskodawca (podmiot odpowiedzialny) do Ministerstwa Zdrowia celem ubiegania się o refundację swojego produktu.
Zgodnie z art. 24, 25 i 25a ustawy refundacyjnej wniosek refundacyjny powinien zawierać szczegółowe dane wnioskodawcy (adres siedziby, dane osoby upoważnionej do reprezentowania firmy w kwestii wniosku), określenie przedmiotu wniosku, dowód dostępności w obrocie leku – czyli kopię decyzji dopuszczenia produktu leczniczego do obrotu, propozycję ceny zbytu i warunki refundacji, szczegółowe informacje o leku (m.in. nazwę, postać, drogi podania, rodzaj opakowania), zobowiązanie do zapewnienia ciągłości dostaw, cenę zbytu, uzyskaną w okresie roku przed złożeniem wniosku, ceny zbytu leku w innych krajach EU i EFTA, dzienny koszt terapii lekiem (dla każdego wskazania oddzielnie), czas trwania standardowej terapii, informacje o ochronie patentowej i wyłączności rynkowej, uzasadnienie wniosku, dowód uiszczenia opłaty za przeprowadzenie weryfikacji refundacyjnej.
I.IV. Uzasadnienie wniosku refundacyjnego
W art. 25 pkt 14 ustawy refundacyjnej znajdujemy wymogi dotyczące treści uzasadnienia wniosku refundacyjnego.
W przypadku leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, który ma co najmniej jeden odpowiednik refundowany w danym wskazaniu uzasadnienie wniosku refundacyjnego powinno zawierać analizę wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych,
Wnioski dotyczące produktów, które nie mają odpowiednika refundowanego w danym wskazaniu powinny natomiast zostać zaopatrzone w analizę kliniczną, sporządzoną na podstawie przeglądu systematycznego w porównaniu z innymi możliwymi do zastosowania w danym stanie klinicznym procedurami medycznymi we wnioskowanym wskazaniu, w tym, o ile występują, finansowanymi ze środków publicznych. W takim przypadku niezbędna będzie także analiza ekonomiczna z perspektywy podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych oraz świadczeniobiorcy oraz analiza wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych. Podmiot składający wniosek powinien przedstawić także w uzasadnieniu analizę racjonalizacyjną, przedkładaną w przypadku gdy analiza wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych wykazuje wzrost kosztów refundacji. Analiza ta powinna przedstawiać rozwiązania dotyczące refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego, wyrobów medycznych, których objęcie refundacją spowoduje uwolnienie środków publicznych w wielkości odpowiadającej co najmniej wzrostowi kosztów wynikającemu z analizy wpływu na budżet,
Zarówno w przypadku produktu, który ma już refundowany odpowiednik, jak i tego, który na liście refundacyjnej znalazłby się po raz pierwszy w uzasadnieniu wniosku powinny znaleźć się informacje dotyczące działalności naukowo-badawczej i inwestycyjnej wnioskodawcy w zakresie związanym z ochroną zdrowia na terytorium Rzeczypospolitej Polskiej oraz w innych państwach członkowskich Unii Europejskiej lub państwach członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA);
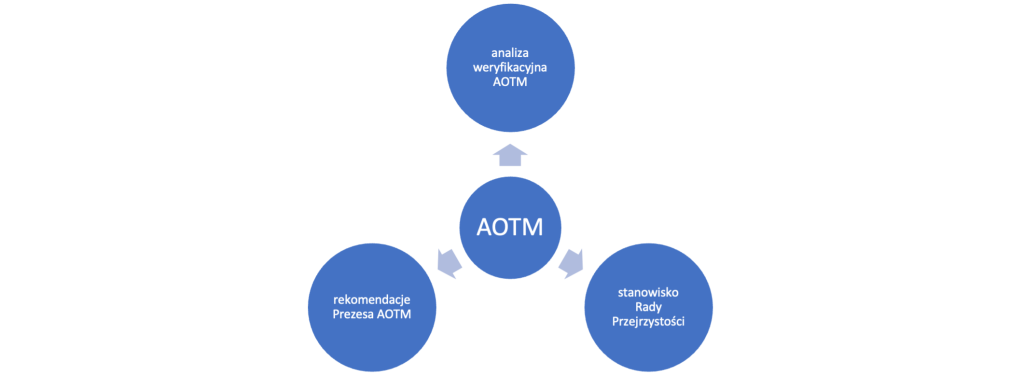
I.V. Ocena wniosku przez Agencję Oceny Technologii Medycznych i Taryfikacji
Wniosek refundacyjny po złożeniu do Ministerstwa Zdrowia przekazywany jest do Agencji Oceny Technologii Medycznych i Taryfikacji (AOTM), gdzie – zgodnie z art. 35 ustawy refundacyjnej, ja jego podstawie przygotowywana jest analiza weryfikacyjna Agencji, stanowisko Rady Przejrzystości oraz rekomendacje Prezesa Agencji.
I.VI. Stanowisko Komisji Ekonomicznej
Komisja Ekonomiczna to zespół, który prowadzi negocjacje cenowe z wnioskodawcami (firmami farmaceutycznymi) w sprawie refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych. Komisja prowadzi negocjacje w składzie pięcioosobowym, zwanym „zespołem negocjacyjnym”.
Zgodnie z art. 17 ust. 2 ustawy refundacyjnej, Komisja liczy 20 członków, w tym 13 przedstawicieli Ministerstwa Zdrowia i 6 przedstawicieli Prezesa Narodowego Funduszu Zdrowia. Od ponad dziesięciu lat, tj. od 13 października 2011 r. przewodniczącym komisji jest prof. Mirosław Szeligowski.
Do zadań Komisji należy prowadzenie negocjacji w zakresie: ustalenia urzędowej ceny zbytu,
poziomu odpłatności, wskazań, w których produkt leczniczy ma być refundowany, monitorowanie realizacji całkowitego budżetu na refundację, racjonalizacja wydatków związanych z refundacją oraz przedstawianie ministrowi zdrowia propozycji w tym zakresie.
Gwarantem niezależności i obiektywności członków Komisji są ograniczenia jakim podlegają: ani sami członkowie, ani ich współmałżonkowie nie mogą pełnić funkcji kierowniczych w spółkach, zajmujących się wytwarzaniem i obrotem produktami leczniczymi. Nie można być także, jednocześnie członkiem Komisji Ekonomicznej i Rady Przejrzystości, ani łączyć członkostwa z funkcją Prezesa Agencji i jego zastępcy.
Po zakończeniu negocjacji następuje wydanie decyzji refundacyjnej przez Ministerstwo Zdrowia, a następnie na tej podstawie lek zostaje wpisany do wykazu leków refundowanych w ramach programów lekowych.
- Postępowanie refundacyjne – jak być powinno?
Już w Polityce Lekowej Państwa na lata 2018-2022 wskazywano na konieczność zwiększenia przejrzystości podejmowanych decyzji refundacyjnych oraz poziomu zaufania w dialogu pomiędzy uczestnikami systemu. Świadczy to o słusznej diagnozie państwa, że proces refundacyjny wymaga diametralnych zmian, które go nie tyle usprawnią, co zracjonalizują. Aktualnie toczą się prace nad projektem nowelizacji ustawy refundacyjnej (projekt w tej chwili jest na etapie prac w Stałym Komitecie Rady Ministrów)[6], który stanowi pierwszą tak poważną próbę zmiany ustawy refundacyjnej. Upływ 11 lat obowiązywania ustawy refundacyjnej pokazał niewątpliwie braki i niedociągnięcia, których ustawodawca nie przewidział w toku prac nad ustawą oraz wady przyjętych rozwiązań. Wiele zaproponowanych zmian wynika z potrzeby doprecyzowania rozwiązań, które obecnie budzą wątpliwości interpretacyjne. Projekt ustawy jest także odpowiedzią na wnioski kierowane do ministra właściwego do spraw zdrowia od przedsiębiorców obecnych na rynku farmaceutycznym, pacjentów i innych grup społecznych, na których funkcjonowanie wpływają przepisy ustawy refundacyjnej[7].
II.I. Propozycje legislacyjne – art. 11 ust. 1a i art. 25 pkt 3
Art. 1 pkt 12 projektu nowelizacji ustawy refundacyjnej przewiduje znaczące zmiany w treści art. 11 i art. 25 ustawy refundacyjnej. Zmiany te mogą znacząco wpłynąć na długość i racjonalność postępowania refundacyjnego.
W myśl projektu nowelizacji do art. 11 w ust. 1 po wyrazie „refundacją” dodaje się wyrazy „lub odmowa objęcia refundacją”, co jest naturalną konsekwencją kolejnej zmiany, tj. po ust. 1 dodaje się ust. 1a w brzmieniu:
„1a. Minister właściwy do spraw zdrowia odmawia wszczęcia postępowania o objęcie refundacją leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, jeżeli w chwili złożenia wniosku o objęcie refundacją co najmniej jeden odpowiednik refundowany w danym wskazaniu posiada ochronę patentową lub ochronę dotyczącą wyłączności rynkowej, o której mowa w art. 15 ust. 2 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne, chyba że wnioskodawca przedłoży dokumenty uprawniające go do prowadzenia obrotu produktem pomimo istnienia wyżej wskazanej ochrony.”
Ponadto art. 25 pkt 3 ustawy refundacyjnej, który obecnie przewiduje, że wniosek o objęcie refundacją musi zawierać „dowód dostępności w obrocie leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, w chwili składania wniosku;”, otrzymać ma brzmienie według którego wniosek refundacyjny winien zawierać:
„dowód dostępności w obrocie leku, środka spożywczego specjalnego przeznaczenia żywieniowego, wyrobu medycznego, na dzień składania wniosku, a w przypadku produktu leczniczego terapii zaawansowanej – zobowiązanie do zapewnienia gotowości technologicznej do jego wytworzenia na dzień składania wniosku;”.
W praktyce dokumentem zaświadczającym o dostępności danego produktu w obrocie na dzień złożenia dla niego wniosku refundacyjnego najczęściej jest obecnie faktura sprzedaży danego produktu do przedsiębiorstwa zajmującego się obrotem hurtowym na terenie Polski. Akceptowane jest także oświadczenie wystawione przez tego przedsiębiorcę o posiadaniu na stanie danej liczby opakowań wnioskowanego produktu.
Proponowane zmiany należy rozpatrywać w kontekście tak zwanych „powiązań patentowych”. Patent linkage oznacza praktykę polegającą na uzależnieniu decyzji o rejestracji lub refundacji leku równoważnego (generyka lub biologicznego równoważnego) od statusu patentowego leku referencyjnego.
Jak wskazuje dr hab. P. Podrecki w swojej opinii do projektu nowelizacji:
„[k]onsekwencją obecnej regulacji jest brak możliwości składania wniosku o refundację generycznego produktu leczniczego przed wygaśnięciem ochrony z tytułu patentu, dodatkowego świadectwa ochronnego bądź wyłączności rynkowej danych rejestracyjnych produktu oryginalnego”[8].
Wymóg zawarty w art. 25 pkt 3 ustawy refundacyjnej uniemożliwia złożenie wniosku refundacyjnego w końcowym okresie ochrony patentowej odpowiednika, przez co opóźnia wejście na rynek tańszego leku o kolejne miesiące po upływie ochrony patentowej.
II.II. Wady i zalety proponowanej nowelizacji
Uzasadnienie do projektu nowelizacji przekonuje, że zmiany w art. 11 ustawy refundacyjnej mają charakter doprecyzowujący obowiązujące przepisy, zaś proponowane dodanie ust. 1a do art. 11 wynika z konieczności zapewnienia transparentności obwieszczeniom refundacyjnym, o których mowa w art. 37 ustawy refundacyjnej.
„Objęcie refundacją danego leku nie może odbywać się w warunkach niepewności, co do możliwości swobodnego nim obrotu na terytorium Rzeczpospolitej Polskiej, z uwagi na fakt, że podmiot starający się o refundację twierdzi, że ochrona patentowa dla leku jego konkurenta jest bezprawna. Z uwagi na negatywne doświadczenia Ministerstwa Zdrowia wynikające ze zdarzeń, które miały miejsce w takich stanach faktycznych zaproponowano regulacje umożliwiające zablokowanie możliwości wydania pozytywnej decyzji dla leku generycznego, którego komparatorem jest oryginał posiadający ochronę prawną wynikającą z patentu lub okresu wyłączności rynkowej. Sytuacja taka jest również niepożądana z punktu widzenia pacjenta, który widzi lek na wykazie leków refundowanych, np. tańszy niż oryginał i nie może go kupić, gdyż jego sprzedaż została zablokowana w postępowaniu sądowym. Proponowane dodanie ust. 1a spowodowane jest koniecznością zapewnienia stabilności list refundacyjnych, w tym podstaw limitów, a tym samym przejrzystością i pewnością obrotu. Brak dotychczas regulacji w tym zakresie powodował, że firmy farmaceutyczne składały wnioski o objęcie refundacją odpowiedników leków występujących w refundacji, pomimo że leki oryginalne posiadały ochronę patentową lub ochronę wynikającą z wyłączności rynkowej. W konsekwencji firmy farmaceutyczne, którym przysługiwały prawa ochronne pozywały firmy naruszające prawa z patentu lub wyłączności rynkowej i uzyskiwały orzeczenia sądowe zakazujące ich konkurentom sprzedaży leku na rynku polskim. W konsekwencji pomimo, że lek widniał na obwieszczeniu refundacyjnym nie mógł być sprzedawany i był niedostępny dla pacjentów. Leki generyczne były oczywiście tańsze niż leki oryginalne, co dla niektórych pacjentów było kwestią przesądzającą o ich nabyciu. Fakt zakazu obrotu takim lekiem na terytorium Rzeczypospolitej Polskiej powodował chaos i liczne problemy oraz dezorientację pacjentów. Stąd konieczne jest uregulowanie wprost tej kwestii przez zobowiązanie ministra właściwego do spraw zdrowia do pozostawienia bez rozpoznania wniosku o objęcie refundacją w sytuacji, gdy wniosek firmy generycznej został złożony w czasie obowiązywania decyzji dla produktu oryginalnego korzystającego z ochrony patentowej lub wyłączności rynkowej”[9].
Argumentacja przedstawiona przez autorów projektu nowelizacji jest co do zasady spójna, klarowna i stanowi próbę racjonalnego rozwiązania problemów jakie zgłaszano do Ministerstwa Zdrowia lub które uwidoczniła praktyka.
Mimo tego należy pamiętać o kluczowym, najważniejszym celu jaki przyświeca całemu przedsięwzięciu jakim jest walka o dostępność, w tym dostępność cenową leków i terapii. Równa możliwość dostępu do leczenia dla wszystkich jest przecież motorem wszelkich działań podejmowanych przez państwo w kierunku usprawnienia i ulepszenia publicznej służby zdrowia i to właśnie ta idea jest fundamentem na którym zbudowane zostały postępowania i procedury refundacyjne. Dlatego też należy pochylić się nad negatywnymi stronami proponowanej nowelizacji, szczególnie w kontekście długości postępowań refundacyjnych (a co za tym idzie okresu czekania na dostępność leku) i proponowanego brzmienia art. 11 ust. 1a.
W przypadku jego uchwalenia art. 11 ust. 1a miałby w praktyce efekt tożsamy z aktualną regulacją ujętą w art. 25 pkt 3, którego treść skutkuje tym, że wniosek o objęcie refundacją generycznego produktu leczniczego można złożyć dopiero po wygaśnięciu ostatniego z reżimów ochronnych (ochrony z tytułu patentu, dodatkowego świadectwa ochronnego bądź wyłączności rynkowej danych rejestracyjnych produktu oryginalnego).
W przypadku wejścia w życie art. 11 ust. 1a w proponowanym brzmieniu złożenie wniosku o objęcie refundacją leku generycznego w trakcie trwania ochrony patentowej lub wyłączności rynkowej obejmującej wnioskowany produkt powodowałoby natomiast wydanie przez Ministra do spraw zdrowia decyzji o odmowie wszczęcia postępowania.
Jak wskazuje w swojej opinii dr hab. P. Podrecki „[p]raktycznego znaczenia nie ma również dodanie do najnowszej propozycji passusu o możliwości przedstawienia dokumentów uprawniających do obrotu produktem będącym przedmiotem ochron. Zawarcie porozumienia
licencyjnego, obejmującego obrót lekiem, pomiędzy podmiotem odpowiedzialnym za lek referencyjny a producentem jago generycznego odpowiednika, w czasie ostatnich miesięcy
trwania prawa wyłącznego jest niezwykle mało prawdopodobne”[10].
Na negatywną ocenę proponowanego art. 11 ust. 1a składa się jednak szereg kwestii z których kluczową z perspektywy pacjenta będzie tzw. farmakoekonomika. Państwo polskie, odpowiadając na słusznie wskazany w Polityce Lekowej Państwa 2018-2022 problem dostępności dla pacjentów nowych leków stanowiących mniej kosztowny zamiennik wobec produktów stosowanych dotychczas, powinno dążyć do maksymalnego zmniejszenia różnicy czasowej pomiędzy wprowadzeniem leku generycznego na rynek a objęciem tego leku refundacją. Znaczna różnica cen pomiędzy lekami oryginalnymi a ich generycznymi odpowiednikami oznaczać będzie znaczące i szybkie obniżenie końcowego kosztu leków refundowanych oraz ich większą dostępność dla pacjentów. Wszak racjonalność wykorzystywania środków publicznych zakłada, iż refundowane powinny być zasadniczo te produkty, które mają udowodnioną skuteczność i bezpieczeństwo stosowania, a relacja efekt terapeutyczny – koszt terapii jest korzystniejszy w porównaniu z dostępnymi wariantami terapii[11].
Art. 11 ust. 1a proponowanej nowelizacji nie tylko nie spełnia założenia reagowania na zdiagnozowany w PLP 2018-2022 problem, co – po ewentualnym jego przyjęciu – będzie go pogłębiać. Ponieważ postępowanie refundacyjne samo w sobie od chwili złożenia wniosku o objęcie refundacją do momentu wydania decyzji o refundacji zajmuje przeciętnie od 6 do nawet 12 miesięcy, a możliwość skutecznego złożenia wniosku o objęcie refundacją generycznego produktu leczniczego pojawi się dopiero po wygaśnięciu ostatniego z reżimów ochronnych leku oryginalnego, nie ma wątpliwości, że w efekcie tej regulacji możliwość refundacji tańszego zamiennika leku oryginalnego zostanie znacznie przesunięta w czasie. Dlatego też należy wycofać się z proponowanej w tym brzmieniu regulacji.
- Propozycje racjonalizujące postępowanie refundacyjne w Polsce
Jak wskazano wyżej, powiązanie patentowe (które występuje już obecnie w ustawie refundacyjnej, a po przyjęciu proponowanej nowelizacji zostanie w niej jeszcze bardziej osadzone) oznacza mechanizm prawny uzależniający uzyskanie decyzji administracyjnoprawnej, z reguły decyzji o dopuszczeniu leku do obrotu lub objęcia do refundacją od jego statusu prawopatentowego (a niekiedy także statusu ochronnego z tytułu praw własności intelektualnej). Status produktu generycznego w odniesieniu do pozwolenia na dopuszczenie do obrotu lub innej decyzji administracyjnej jest więc powiązany ze statusem patentu lub innych reżimów ochronnych dotyczących referencyjnego produktu leczniczego.
III.I. Eliminacja powiązań patentowych jako element racjonalizujący proces refundacyjny
Patent linkage to mechanizm budzący wiele wątpliwości. Uznaje się go za sprzeczny z wymaganiami racjonalnej farmakoekonomiki systemu refundacyjnego – wszak refundowane powinny być te produkty, które mają udowodnioną skuteczność i bezpieczeństwo stosowania, przy czym obejmowanie refundacją leku oryginalnego w sytuacji, gdy na rynku dostępny jest jego tańszy odpowiednik jest po prostu nieracjonalnym wykorzystywaniem środków z budżetu refundacyjnego. Racjonalne postępowanie refundacyjne powinno uwzględniać nie tylko efekt terapeutyczny, ale i koszt terapii danymi produktami. Rozwiązanie w myśl którego złożenie wniosku refundacyjnego jest możliwe dopiero w dniu następnym po ustaniu ochrony w odniesieniu do leku oryginalnego sprawia, że przez kilka miesięcy (średnio 6-8 miesięcy trwania procedury refundacyjnej) od wprowadzenia leku generycznego do obrotu, na rynku dostępna jest terapia alternatywna wobec terapii lekiem oryginalnym, o takim samym stopniu skuteczności i bezpieczeństwa, jednak znacznie tańszym potencjalnym koszcie refundacji, co w zupełności nie jest wykorzystywane przez państwo, które powinno dbać o racjonalność wydatkowania środków budżetowych, szczególnie w kwestiach związanych ze zdrowiem.
Należy także zwrócić uwagę na fakt, że mechanizm powiązania patentowego jest szeroko kwestionowany jako niezgodny z prawem konkurencji i niespójny z prawem farmaceutycznym oraz prawem własności intelektualnej. Państwo powinno dążyć do eliminowania patent linkage, tymczasem proponowana nowelizacja ustawy refundacyjnej idzie w kierunku przeciwnym. Już obecnie obowiązujące powiązanie patentowe zawarte w art. 25 pkt 3 ustawy refundacyjnej powoduje nieuzasadnione blokowanie refundacji odpowiedników leków oryginalnych, a co za tym idzie utrzymywanie się wyższych cen produktów, czyli dodatkowe wydatki na refundację. Materiałem na odrębne opracowanie jest w tym przypadku także kwestia ochrony konkurencji, która jest w tym przypadku znacznie zagrożona.
Powiązania patentowe w świetle unijnych zasad prawa konkurencji są mechanizmem bardzo wątpliwym. Prawo unijne nie pozwala na powiązanie pozwolenia na dopuszczenie do obrotu ze statusem patentu na monopolistyczny produkt referencyjny. Zgodnie z art. 81 Rozporządzenia (WE) 726/20045 Parlamentu Europejskiego i Rady z dnia 31 marca 2004 r.[12] i art. 126 Dyrektywy 2001/83/WE6 Parlamentu Europejskiego i Rady z dnia 6 listopada 2001 r.[13] organy odpowiedzialne za pozwolenia na dopuszczenie do obrotu nie powinny brać pod uwagę statusu patentowego oryginalnego produktu referencyjnego przy rozpatrywaniu wniosku o pozwolenie na dopuszczenie do obrotu generycznych produktów leczniczych. Pozwolenie na dopuszczenie do obrotu produktu leczniczego jest wydawane na podstawie kryteriów naukowych dotyczących jakości, bezpieczeństwa i skuteczności danego produktu leczniczego: kryteria te są związane ze względami zdrowia publicznego. Nie są nimi natomiast kryteria związane z podleganiem ochronie własności intelektualnej, których ważność lub naruszenie można rozpatrywać jedynie na przed właściwymi sądami.
- Podsumowanie
Funkcjonowanie powiązań patentowych w systemie refundacyjnym skutkuje licznymi negatywnymi konsekwencjami, w tym na polu konkurencji firm farmaceutycznych – kluczowe pozostaje jednak opóźnianie faktycznej dostępności leku, który jest odpowiednikiem leku dotychczas dostępnego i refundowanego, a który ma tożsame wartości terapeutyczne, jednakże jego cena jest niższa w stosunku do ceny produktu oryginalnego. Brak reakcji ze strony państwa na tak nieracjonalne wydatkowanie środków budżetowych, a wręcz projektowanie zmian, które taki stan pogłębią wymaga sprzeciwu i zmiany polityki w kwestii powiązań patentowych.
Mechanizm umożliwiający występowanie z wnioskiem o decyzję refundacyjną w ciągu określonego czasu przed upływem ostatniego z reżimów ochronnych, obejmujących dany produkt leczniczy zaproponowany w opinii do projektu nowelizacji autorstwa dr hab. P. Podreckiego[14] wydaje się być najbardziej racjonalnym rozwiązaniem omawianego problemu. Taka formuła pozwoli zniwelować lub znacznie ograniczyć negatywne konsekwencje mechanizmu powiązań patentowych, przy jednoczesnym uwzględnieniu motywów zamieszczonych w uzasadnieniu do projektu nowelizacji, które stały za przedstawieniem projektowanego art. 11 ust. 1a w analizowanym brzmieniu.
[1] Konstytucja Rzeczypospolitej Polskiej z dnia 2 kwietnia 1997 r. (Dz.U. z 1997 r. nr 78, poz. 483, z 2001 r. nr 28, poz. 319, z 2006 r. nr 200, poz. 1471, z 2009 r. nr 114, poz. 946), dalej jako: Konstytucja RP.
[2] Ministerstwo Zdrowia, Polityka Lekowa Państwa 2018-2022, s. 66.
[3] Ibidem, s. 65.
[4] Art. 2 pkt 26 ustawy z dnia 12 maja 2011 r. o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych (Dz. U. z 2022 r., poz. 2555 tj.j. z późn. zm.), dalej jako: „ustawa refundacyjna”.
[5] Zob. Obwieszczenie Ministra Zdrowia z dnia 21 grudnia 2022 r. w sprawie wykazu refundowanych leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych na 1 stycznia 2023 r., https://www.gov.pl/web/zdrowie/obwieszczenie-minister-zdrowia-z-dnia-21-grudnia-2022-r-w-sprawie-wykazu-refundowanych-lekow-srodkow-spozywczych-specjalnego-przeznaczenia-zywieniowego-oraz-wyrobow-medycznych-na-1-stycznia-2022-r, (dostęp: 31.12.2022).
[6] Projekt z dnia 13.10.2022: Projekt ustawy o zmianie ustawy o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych oraz niektórych innych ustaw, https://legislacja.rcl.gov.pl/projekt/12348505/katalog/12799519#12799519, (dostęp: 31.12.2022), dalej jako: „projekt nowelizacji”.
[7] Zob. Uzasadnienie do projektu nowelizacji w wersji z dnia 13.10.2022, https://legislacja.rcl.gov.pl/projekt/12348505/katalog/12799519#12799519, (dostęp: 31.12.2022).
[8] P. Podrecki, Opinia prawna dotycząca proponowanego art. 11 ust. 1a projektu nowelizacji Ustawy o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych (Dz. U. 2011 Nr 122, poz. 696 z późn. zm.) z dn. 30.06.2021 r., https://www.producencilekow.pl/wp-content/uploads/2022/12/Pismo-PZPPF-W.467.2022-za%C5%82%C4%85cznik_opinia-prawna-prof.-Podreckiego-na-temat-art_11_ust_1a1.pdf, (dostęp: 30.12.2022), s. 3.
[9] Uzasadnienie projektu nowelizacji, s. 13-15.
[10] Op. cit. P. Podrecki, Opinia…, s. 3.
[11] Zob. Ustawa o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych. Komentarz (red.) M. Pieklak, R. Stankiewicz, M. Czarnuch, M. Mądry, 2014.
[12] Rozporządzenie (WE) Nr 226/2004 Parlamentu Europejskiego i Rady z dnia 31 marca 2004 r. ustanawiające unijne procedury wydawania pozwoleń dla produktów leczniczych stosowanych u ludzi i do celów weterynaryjnych i nadzoru nad nimi oraz ustanawiające Europejską Agencję Leków (Dz.U. L 136 z 30.4.2004, s. 1), https://eur-lex.europa.eu/legal-content/PL/TXT/PDF/?uri=CELEX:02004R0726-20190128&from=EN, (dostęp: 31.12.2022).
[13] Dyrektywa 2001/83/WE Parlamentu Europejskiego i Rady z dnia 6 listopada 2001 r. w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi (Dz.U. L 311, 28/11/2001 P. 0067-0128), https://eur-lex.europa.eu/legal-content/PL/TXT/?uri=celex%3A32001L0083, (dostęp: 30.12.2022).
[14] Op. cit. P. Podrecki, Opinia…, s. 14
Czytaj także
- The pitfalls of climate & energy policy in the 21st century
- Światowa Organizacja Zdrowia – zastrzeżenia w kontekście aktualnych projektów legislacyjnych
- Dokąd zmierza Europa? Czy planowane zmiany traktatowe to koniec tego, co zostało z suwerenności państw członkowskich Unii Europejskiej?
- Inwestowanie w Fundusze ESG
- A proposal for a EU-wide shared heritage promotion programme
- FinTech – the Future of Europe’s Banking System?
- Raportowanie niefinansowe ESG – część 2